熱線:021-66110810,66110819
手機:13564362870
熱線:021-66110810,66110819
手機:13564362870
能量應激反應途徑。
為了測試SDS細胞利用哪些代償機制來抵消能量生產缺陷(能量應激),我們評估了AMP激活蛋白激酶(AMPK)和PI3K/AKT/哺乳動物雷帕霉素靶標(mTOR)通路,它們是能量缺乏激活的主要調節機制。
在應激條件下,AMPK通過AMP積累被激活,拮抗mTOR并刺激糖酵解等替代性分解代謝過程,從而抵消能量限制。PI3K/AKT/mTOR通路通過線粒體和核糖體的生物生成誘導細胞增殖。被PI3K(磷脂酰肌醇3-激酶)在Thr803處磷酸化的AKT會抑制AMPK并誘導mTOR激活。因此,在野生型細胞中,當AMPK被激活時,PI3K/AKT/mTOR通路通常會受到抑制。
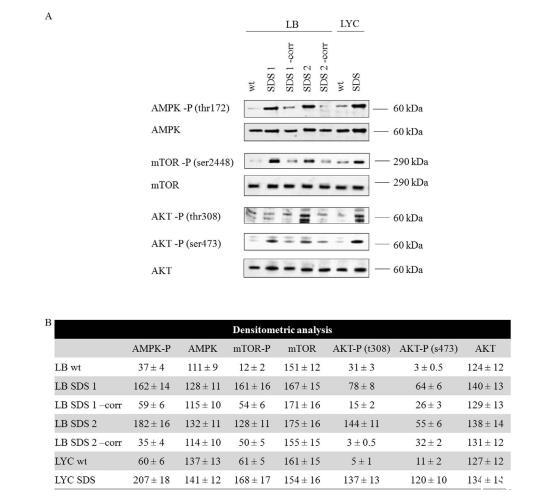
如預期的那樣,在SDS細胞中,AMPK比對照組更活躍,如蛋白磷酸化形式的Western印跡分析所示(圖3)。令人驚訝的是,我們發現與正常對照組和校正對照組相比,mTOR并未受到抑制,反而高度磷酸化。
與這一發現相一致的是,我們發現與野生型細胞和校正細胞相比,SDS細胞中AKT在Thr803和Ser473處也過度磷酸化,從而證實了整個PI3K/AKT/mTOR通路的過度激活。
總之,這些實驗表明,SDS細胞對高能應激反應異常,而這種反應與SDS蛋白有關,因為在校正細胞中未見PI3K/AKT/mTOR通路超活化。
SDS細胞的細胞質鈣濃度水平很高。
Ca2+調節真核蛋白質翻譯和許多其他消耗ATP的細胞反應,因此是蛋白質合成和能量代謝的重要信號分子。此外,高[Ca2+]i與ROS細胞毒性、脂質過氧化和OXPHOS功能密切相關,并且會抑制復合體IV的活性。
在靜息狀態下,SDS細胞的[Ca2+]i比野生型細胞(65±2nM)或校正細胞(64±1nM)高出兩倍(133±8nM)(表1)。
為了研究[Ca2+]i增高的原因,我們評估了ER從細胞質中捕獲并儲存鈣的能力。事實上,ER是細胞內主要的鈣儲存場所,并通過SERCA(鈣ATP酶通道)活性在Ca2+穩態中發揮重要作用,SERCA可將Ca2+從細胞質轉移到ER。硫代碳酸氫鹽(TG)不可逆地阻斷SERCA通道,導致Ca2+從ER泄漏,從而增加[Ca2+]i。與SDS細胞相比,高劑量TG處理(3μM)會導致野生型細胞和校正細胞中的[Ca2+]i顯著增加,從而表明這些細胞在ER中儲存Ca2+的能力受損,這可能是[Ca2+]i增加的原因(表1)。
盡管我們沒有發現任何SERCA通道活性差異,但我們推測[Ca2+]i的增加可能取決于我們在SDS細胞中觀察到的mTOR激活,已知mTOR可通過肌醇-1,4,5三磷酸受體正向調節鈣釋放。
亮氨酸可恢復正常代謝表型。
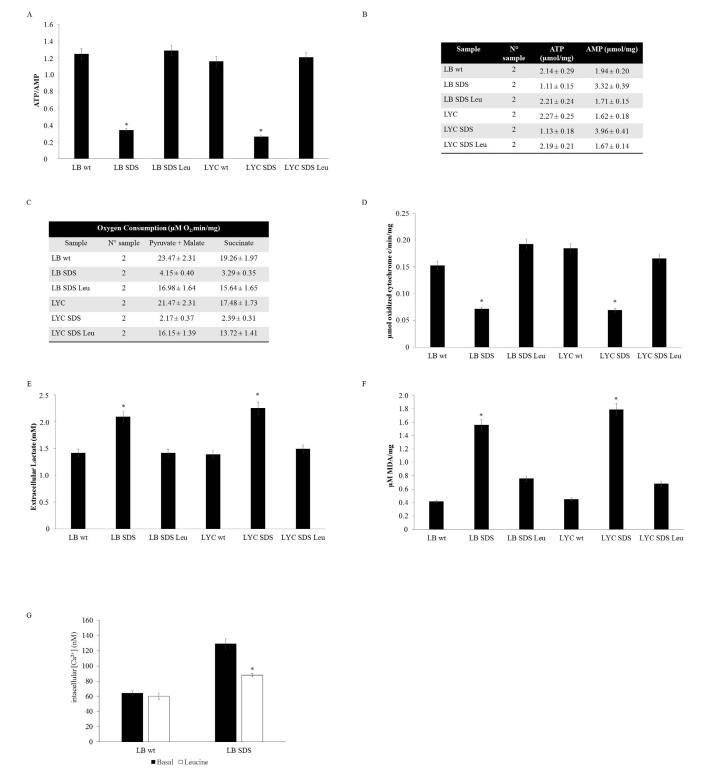
由于亮氨酸(Leucine,Leu)是一種已知能促進蛋白質合成的必需氨基酸,即使在SBDS缺乏的細胞中也是如此,因此我們測試了亮氨酸在處理5天后對SDS淋巴細胞和淋巴母細胞的生化影響。經過Leu處理后,復合體IV的功能得到恢復,呼吸速率和ATP/AMP比率與對照組相當(圖4A-D),細胞內鈣濃度也有所降低(圖4G)。表型逆轉還與脂質過氧化和乳酸產生的減少有關(圖4E、F)。令人驚訝的是,用N-乙酰半胱氨酸(NAC)(一種抗氧化分子,是還原型谷胱甘肽的前體)處理后,在SDS細胞中觀察到的呼吸缺陷、能量窘迫和鈣平衡改變沒有恢復或恢復較差。
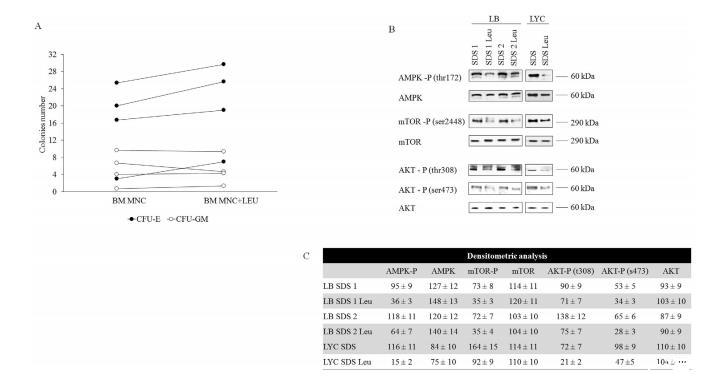
然后,我們測試了Leu對SDS患者骨髓造血干細胞生長的影響,觀察到紅細胞集落生長適度增加,但髓系細胞集落生長沒有增加(圖5A)。由于Leu是mTOR激活的調節劑,我們研究了它在慢性SDS細胞處理后對能量應激途徑的影響。不出所料,AMPK磷酸化的減少是正常OXPHOS恢復的結果。有趣的是,Leu處理后AKT和mTOR磷酸化水平也降低了(圖5B)。可以推測這些影響是由于依賴于營養過剩的AKT/mTOR通路的反饋調節所致。說明Leu恢復了SDS細胞的生化表型,該分子可被視為恢復SDS細胞能量代謝的潛在工具。
討論
SBDS蛋白在核糖體生物生成和蛋白質合成過程中發揮作用,而這兩個高耗能過程與細胞能量的產生密切相關。細胞呼吸是將營養物質轉化為生化能量(主要是ATP)的一系列代謝反應和過程。這一過程由一系列稱為電子傳遞鏈的蛋白質復合物完成,電子傳遞鏈位于線粒體內膜,并與線粒體膜間隙和基質相連。呼吸功能受損會影響ATP的產生,并使細胞面臨能量和氧化壓力。
在本研究中,我們評估了SDS細胞的能量代謝,發現在丙酮酸/蘋果酸或琥珀酸的誘導下,氧消耗會受損。因此,ATP生成減少,AMP積累,從而改變了ATP/AMP比率。
電子通過兩條途徑傳輸。第一種由復合物I、III和IV組成,從NADH中轉移電子,可由丙酮酸/蘋果酸誘導。另一種需要復合體II、III和IV,由琥珀酸激活,從FADH2中轉移電子,能量產生效率低于第一種途徑。由于丙酮酸/蘋果酸和琥珀酸在SDS細胞中對耗氧量的影響程度相同,我們對兩種途徑共有的兩個復合物(III和IV)的活性進行了研究,結果表明復合物IV不能正常工作。
復合體IV活性受損的原因尚不清楚。我們評估了分別由核基因和線粒體基因編碼的復合體IV的兩個亞基COX5A和COX2的表達水平,發現這兩種蛋白的表達水平正常。這表明,盡管SDS細胞中存在核糖體生物發生和轉導缺陷,但它們的合成并沒有受損。不過,值得注意的是,復合體IV由32個蛋白質組成,其中3個由線粒體DNA編碼,29個由核DNA編碼。此外,其中11個是結構蛋白,18個是組裝因子;因此,我們不能排除其他亞基表達較少或折疊不正確的可能性。
另一個原因可能是線粒體膜發生了變化,這是由復合體III和IV之間或復合體IV與氧之間的電子傳遞發生變化所決定的,從而導致細胞色素c氧化酶活性受損,氧化應激增加。然而,這種可能性似乎不大,因為我們在SDS細胞中測得的氧化應激水平較低,并觀察到線粒體膜明顯完整。
值得注意的是,盡管復合體IV在ROS生成中起間接作用,但復合體IV的損傷可能是由復合體I和復合體III的上游電子累積引起的,從而導致ROS生成增加。事實上,線粒體電子傳遞鏈包含多個氧化還原中心,可直接將電子傳遞給氧氣,從而產生ROS。不過,也可能有其他結構參與了氧化應激誘導。特別是,由于SDS細胞的特點是蛋白質生物生成缺陷,缺陷蛋白質的積累可能會導致內質網中的未折疊蛋白反應。
復合體IV受損的另一個可能原因與所報道的SDS細胞中鈣水平的改變有關。細胞內的鈣平衡在調控生化通路中發揮著重要作用,而生化通路可調節對高能應激的反應。特別是,Ca2+對能量代謝有雙重影響。一種作用是,[Ca2+]i可增強OXPHOS,從而激活克雷布斯循環脫氫酶和線粒體底物轉運體。另一個作用是,[Ca2+]i可能通過競爭復合體IV的陽離子結合位點而影響其活性。
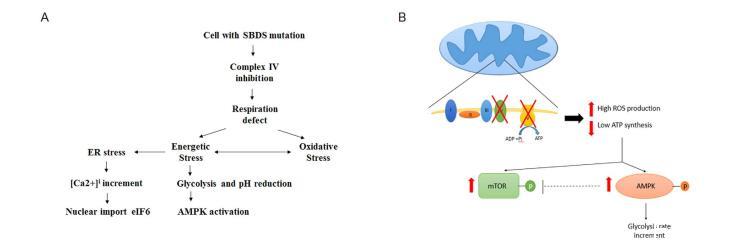
考慮到這些數據(圖6),在SDS細胞中觀察到的高[Ca2+]i可能與解釋SDS細胞的生化表型有關。SBDS與類延伸因子1(EFL1)合作,使eIF6從前60S核糖體亞基中釋放,從而形成核糖體80S。因此,在SDS細胞中,60S亞基的成熟是有缺陷的,其與40S亞基的結合也受到損害。此外,即使在細胞質中,eIF6與前60S亞基的結合也會持續存在,阻止其循環到核仁。考慮到eIF6的核導入受細胞內Ca2+的調節,我們可以推測,SDS細胞中細胞質鈣濃度的增加試圖平衡核糖體生物發生的缺陷。
能量應激會誘導細胞新陳代謝發生變化,從而刺激或抑制參與能量平衡調節的分子網絡,如AMPK和mTOR。在觀察到的SDS細胞中,由于能量應激,AMPK被過度激活,糖酵解途徑受到刺激。令人驚訝的是,我們還發現AKT/mTOR通路被異常地過度激活,因為這兩種蛋白都被過度磷酸化。我們推測,mTOR過度激活是SDS細胞支持能量缺陷和蛋白質合成的一種方式,目的是改善OXPHOS活性。然而,用mTOR拮抗劑雷帕霉素處理SDS細胞可完全抑制殘余的OXPHOS活性,從而減少ROS的產生,進而通過糖酵解恢復能量。這表明,在SDS細胞生化改變的過程中,OXPHOS活性的增加在保證ATP產量增加的同時,也可能導致氧化應激的增強和細胞損傷的潛在增加(圖6)。在AMPK激活的情況下,mTOR激活的另一個潛在機制可能與自噬激活有關。在核糖體病中,氧化應激的增加會通過mTOR-S6激酶途徑誘導自噬。考慮到本手稿中報告的數據,我們可以假設AMPK活性增強可能與細胞能量狀態受損有關,而mTOR通路激活可能與氧化應激產生水平較高有關。
此外,mTOR還控制著幾個依賴Ca2+的過程。例如,它正向調節IP3R(三磷酸肌醇受體)介導的Ca2+釋放,并與Akt共同參與調節線粒體相關內質網膜(MAM)的完整性、鈣通量和能量代謝。
最后,糖酵解代謝的增加會引起乳酸的積累和細胞內pH值的降低,從而導致鈣泵受到抑制,細胞膜[Ca2+]隨之增加(圖6)。
亮氨酸是一種必需氨基酸,能促進細胞增殖和蛋白質合成。核糖體病或其他核糖體生物發生缺陷的病癥可從這種氨基酸的治療中獲益。與這些研究一致,我們發現亮氨酸能改善SDS患者的體外紅細胞生成。此外,用亮氨酸處理SDS細胞可恢復OXPHOS和ATP合成,降低細胞質鈣濃度以及AMPK和AKT/mTOR活性,這表明亮氨酸可能有助于維持SDS患者紊亂的能量代謝和紅細胞生成。特別是,考慮到亮氨酸能提高OXPHOS活性,部分恢復呼吸作用,因此它可能是AMPK和AKT/mTOR通路的調節劑,能恢復它們的生理作用。我們可以推測,亮氨酸誘導的氧化應激減少會導致mTOR-S6激酶途徑失活。
最后,值得注意的是,能量代謝是維持自我更新干細胞的決定因素。骨髓中的造血干細胞被封閉在缺氧的微環境中。缺氧條件使造血干細胞處于靜止狀態,這與糖酵解代謝有關。在(不對稱)自我更新干細胞分裂過程中,一個細胞保持干細胞的糖代謝特征,而另一個細胞則進入血管,獲得OXPHOS代謝并進行分化。造血干細胞對氧化應激增加很敏感,而線粒體氧化磷酸化是產生ROS的主要原因。因此,糖酵解代謝是造血干細胞自我更新維持的決定因素。為了減少線粒體代謝,造血干細胞會積極調節AMPK,同時抑制PI3K/mTOR通路。AKT或mTOR信號的持續活躍和高[Ca2+]i水平會導致增殖增加和造血干細胞貧化。因此,這項研究可為了解導致SDS患者骨髓衰竭的生化途徑提供一個新的視角。
總之,我們首次發現,盡管SDS細胞的線粒體形態沒有發生顯著變化,但它卻遭受著與SBSD蛋白缺陷有關的能量應激和嚴重的呼吸缺陷。這些缺陷可通過增強的AMPK、糖酵解和mTOR/Akt通路激活得到部分補償。在維持這種改變的新陳代謝中起關鍵作用的可能是鈣平衡的改變。
 相關新聞
相關新聞